




随着人类基因序列的完成,蛋白质组学热浪掀起了后基因组时代的序幕。蛋白质组学是以蛋白质组为研究对象的新研究领域,主要研究细胞内蛋白质的组成及其活动规律,建立完整的蛋白质文库。蛋白质组学与基因组学同等重要,甚至更为重要,因为基因的重要作用最终是由蛋白质来体现。后基因时代科学家们志在盘点人类蛋白质组内所有的蛋白质,研究其生理功能。然而由于人体存在的20300个基因,编码着数百万种不同的蛋白质分子,其复杂性使得目前全面绘制人类蛋白质组图谱成为了一项非常艰巨的任务。
近日由美国伊利诺大学和西北大学化学生物学家Neil Kelleher领导的一个研究小组开发了一项新技术,利用这一技术研究人员快速分离和鉴别了数千个完整的蛋白质分子。这一研究发现对推动科学家们解析疾病机制,实现早期诊断疾病及靶向性治疗具有重要的意义。这一研究成果于10月30日在线发表在《自然》(Nature)杂志上。
蛋白质的基本结构是氨基酸序列,通过鉴定氨基酸序列来匹配它的蛋白质,这是定性研究,是蛋白质组学研究的基础。现在蛋白质组学研究已达到的基本研究手段是:以生物质谱技术为核心,对蛋白质进行大规模、高通量分离、鉴定和分析。以生物质谱为基础的蛋白质组学分析方法主要有Bottom-up和Top-down两种方法。
Bottom-up(自下而上)是一种传统的手段,它将蛋白质的大片段混合物消化/酶解成小片段的肽后再进行分析,是在蛋白质组学的研究中较为广泛使用的一种质谱技术。然而由于选择性剪接、各种蛋白质修饰(例如乙酰化和甲基化)以及内源性蛋白质裂解等复杂机制的存在,使得细胞内产生了复杂的蛋白异构体及种类。Bottom-up无法完整准确地鉴定这些蛋白的特征。
Top-down(自上而下)技术虽可以直接对完整的蛋白——包括翻译后修饰蛋白以及其它一些大片段蛋白测序,然而由于不能将完整蛋白质片段分离技术与串联质谱技术相整合而无法开展大规模的蛋白质组研究。
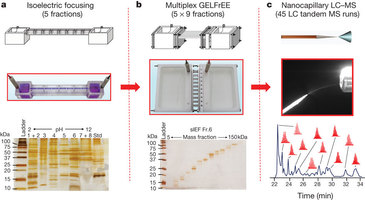
在这篇文章中,研究人员开发出了一个新型的四维分离系统(four-dimensional separation system),利用它鉴别了人类细胞中1043个基因编码,在经过翻译后修饰(PTEM)、RNA剪接和蛋白质水解作用后生成的3000多种蛋白质。这一系统将蛋白质的分离效率及蛋白质组的覆盖率提高了20倍,从而鉴别出了105 kDa大小,包含11个跨膜螺旋的蛋白。此外,还检测出了许多从前难以发现的内源性人类蛋白异构体。结合最新的Swiss-Prot数据库数据,研究人员揭示出了个别基因和全部蛋白质分子之间的相互关系。
“分析细胞中表达的全部蛋白质对于蛋白质组学领域来说是一个重大的技术挑战,”美国国立卫生研究所下属国家常规医学科学研究所蛋白质组研究基金负责人Charles Edmonds说:“Kelleher博士及同事利用多重分离与质谱分析法相结合的新技术将蛋白质组的覆盖率提高了超过一个数量级,这标志蛋白质组学研究一个新的开端。”
Mapping intact protein isoforms in discovery mode
using top-down proteomics
Abstract A full description of the human proteome relies on the challenging task of detecting mature and changing forms of protein molecules in the body. Large-scale proteome analysis1 has routinely involved digesting intact proteins followed by inferred protein identification using mass spectrometry2. This ‘bottom-up’ process affords a high number of identifications (not always unique to a single gene). However, complications arise from incomplete or ambiguous2 characterization of alternative splice forms, diverse modifications (for example, acetylation and methylation) and endogenous protein cleavages, especially when combinations of these create complex patterns of intact protein isoforms and species3. ‘Top-down’ interrogation of whole proteins can overcome these problems for individual proteins4, 5, but has not been achieved on a proteome scale owing to the lack of intact protein fractionation methods that are well integrated with tandem mass spectrometry. Here we show, using a new four-dimensional separation system, identification of 1,043 gene products from human cells that are dispersed into more than 3,000 protein species created by post-translational modification (PTM), RNA splicing and proteolysis. The overall system produced greater than 20-fold increases in both separation power and proteome coverage, enabling the identification of proteins up to 105 kDa and those with up to 11 transmembrane helices. Many previously undetected isoforms of endogenous human proteins were mapped, including changes in multiply modified species in response to accelerated cellular ageing (senescence) induced by DNA damage. Integrated with the latest version of the Swiss-Prot database6, the data provide precise correlations to individual genes and proof-of-concept for large-scale interrogation of whole protein molecules. The technology promises to improve the link between proteomics data and complex phenotypes in basic biology and disease research7.
Nature
原文链接:http://www.nature.com/nature/journal/vaop/ncurrent/full/nature10575.html